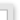| 結構式 |
|
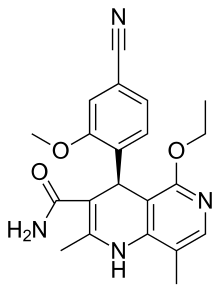
Finerenone
(4S)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-
dimethyl-1,4-dihydro-1,6-naphthyridine-3-carboxamide。
|
| UpToDate |
UpToDate 連結
|
| 藥理作用 |
|
|
Finerenone 是礦物性皮質素受體(MR) 的非類固醇選擇性拮抗劑,MR 可被醛固酮和皮質醇活化並調節基因轉錄。Finerenone 在上皮( 例如腎臟) 和非上皮( 例如心臟和血管) 組織中阻斷MR 介導的鈉再吸收和MR 過度活化。MR 過度活化被認為與纖維化和發炎有關。Finerenone 對MR 具有很高的效價和選擇性,對雄性素、黃體素、雌激素和糖皮質素受體則沒有相關親和力。
|
| 適應症 |
|
|
用於第二型糖尿病 (T2D)相關的慢性腎臟病(CKD)的成年病人,可降低持續性腎絲球過濾率(eGFR)下降、末期腎病(ESKD)、心血管死亡、非致命性心肌梗塞以及因心衰竭住院的風險。
|
| 用法用量 |
|
根據eGFR判定Kerendia 的建議起始劑量,eGFR ≥60: 每天1次20毫克,eGFR ≥25至<60: 每天1次10毫克,eGFR <25: 不建議使用。
無法以口吞服整粒藥錠的病人,本藥品可磨碎後與水或軟質食物( 如蘋果泥) 混和後立即服用。
|
| 藥動力學 |
|
吸收
Finerenone 口服後會完全吸收, 但經過代謝後, 絕對生體可用率為44%。Finerenone 在給藥後的0.5 至1.25 小時達到Cmax。
食物的影響
與高脂肪、高熱量食物一起給藥後,對finerenone 的AUC 沒有臨床顯著影響。
分佈
Finerenone 在穩定期的分佈體積(Vss) 為52.6 L。Finerenone 在體外的血漿蛋白結合率為92%,主要是與血清白蛋白結合。
排除
終端半衰期約為2 至3 小時,全身血液清除率約為25 L/h。
代謝
Finerenone 主要由CYP3A4 代謝(90%),少部分由CYP2C8 (10%) 代謝為無活性代謝物。
排泄
大約80% 的給藥劑量透過尿液排泄(< 1% 為原形) 和約20% 經糞便排泄(< 0.2%為原形)。
|
| 副作用 |
|
|
高血鉀。
|
| 禁忌 |
|
Kerendia 禁用於以下病人:
1. 病人接受強效CYP3A4 抑制劑併用治療,例如: Itraconazole、ketoconazole、ritonavir、nelfinavir、cobicistat、clarithromycin、telithromycin、nefazodone。
2. 患有腎上腺功能不全。
|
| 注意事項 |
|
高血鉀症
Kerendia 可能導致高血鉀症。發生高血鉀症的風險隨著腎功能下降而增加,並且在基期鉀濃度較高或其他有高血鉀症風險因子病人中的風險更高。開始Kerendia 治療前測量所有病人的血清鉀濃度和eGFR 並相應給藥。若血清鉀> 5.0 mEq/L,請勿開始使用Kerendia;若病人血清鉀濃度> 4.8 至5.0 mEq/L,則可考慮開始Kerendia 治療,並根據病人特徵以及血清鉀濃度在開始治療的4 週內進行額外血清鉀監測。
|
| 過量處理 |
|
如果懷疑藥物過量,請立即停止Kerendia 的治療。藥物過量的最可能的表現是高血鉀症。如果出現高血鉀症,應開始標準治療。
由於Finerenone 與血漿蛋白結合的比例約為90%,因此finerenone 不太可能通過血液透析有效移除。
|
| 藥品保存方式 |
|
|
請儲存於30°C以下。
|